

Accurate Single cell Genomic Analysis Holds the Key to Understanding Cancer Heterogeneity
Advances in genomic technologies have significantly improved our understanding of cancer heterogeneity. Single cell whole genome sequencing enables the genetic characterization of distinct tumor cells, providing actionable information for treatment decisions and disease management. In this Application Note, we discuss a novel and accurate method for single cell whole genome amplification Primary Template directed Amplification (PTA) and provide examples of how it supports the detection of low frequency variants in populations of cells.
Cancer heterogeneity drives outcomes
In 2020, 10 million people lost their lives to cancer, and twice as many new cases were reported worldwide.1 These numbers are staggering, despite 50 years of intense global research.
Key insights into the heterogeneous nature of cancer on both a cellular and molecular level came from Peter Nowell's 1976 "clonal theory of cancer" (Figure 1), which laid the foundation for improved


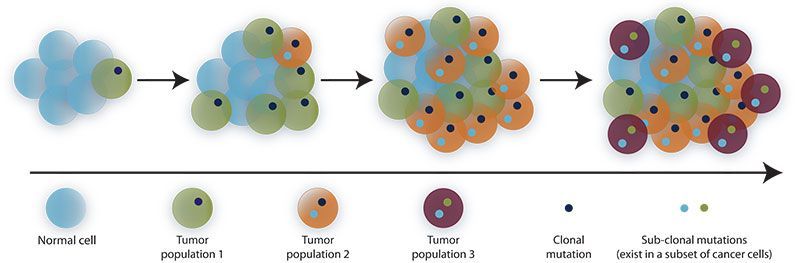
Figure 1. The clonal theory of cancer states that tumor initiation occurs by an induced change in a single, previously normal cell which makes it "neoplastic" and provides it with a selective growth advantage over adjacent normal cells. Mutations that favor immune escape, tumor survival, metastasis, and therapy resistance arise continually, persist in small cell populations, and accumulate over time.
disease management and modern, targeted therapies.2 Since then, ever advancing genomic technologies have unlocked the genetic basis of cancer at increasing resolution. Nevertheless, most of our knowledge derives from bulk omics approaches. Analysis of mixed cell populations masks the complex heterogeneity within and across tumors, revealing only the most abundant variants in a sample. The promise of precision medicine will only be realized through routine genomic analysis of single pathogenic cells, which allows for extremely sensitive detection of conserved, rare and de novo variant alleles. To date, widespread adoption of this approach has been hampered due to the limitations of existing single cell whole genome amplification (WGA) methods.
PTA enables accurate single cell genomics
Primary Template directed Amplification (PTA) is a novel, isothermal method that employs innovative chemistry to drive whole genome amplification of ultra low quantities of DNA from the primary template. This significantly reduces challenges associated with existing WGA approaches (such as variable coverage and allelic dropout), and enables the detection of single nucleotide variants (SNVs), translocations, and copy number variation (CNV) from a single assay.3 For Research Use Only. Not for use in diagnostic procedures.
To demonstrate the ability of PTA to discern discrete SNVs at single cell resolution, we induced resistance in the AML cell line MOLM 13 through exposure to the tyrosine kinase inhibitor quizartinib. Whole genome sequencing (WGS) of individual parental and resistance cells confirmed that single cell analysis significantly increases variant allele frequency (VAF) sensitivity (Figure 2). In an additional study, CNV analysis of a well characterized, hypertriploid breast cancer cell line (SKBR3) was performed. CNV heterogeneity (not distinguishable in a bulk sample) was clearly observed between individual single cells (Figure 3). In addition, PTA supports the analysis of single cell tumor mutation burden (TMB).4
Conclusion
BioSkryb’s ResolveDNA® Whole Genome Sequencing Workflow employs PTA to enable accurate and scalable single cell sequencing. Our BaseJumper™ Bioinformatics Platform is designed to sort, analyze and interpret very large data sets. This novel data collection and processing platform provides an order of magnitude of improvement in sensitivity over conventional bulk sequencing methods.

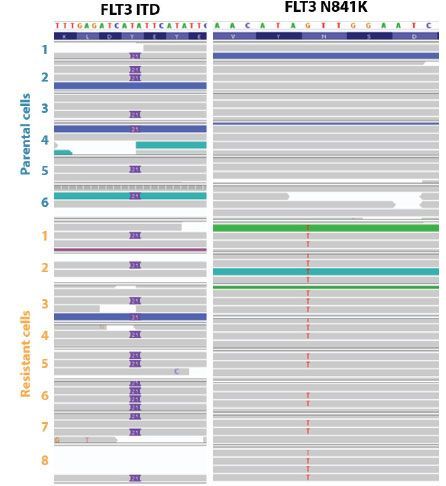
Figure 2. Digital detection of somatic alleles in single cells. Parental and quizartinib-resistant AML cells were both found to contain the FLT3 internal tandem duplication (FLT3 ITD, left). An additional, secondary, missense mutation (N841K) was detected in resistant cells only (right). This mutation, which is likely a key driver of drug resistance, emerged as dominant in resistant cells, after being rare in the parental population (confirmed by qPCR).

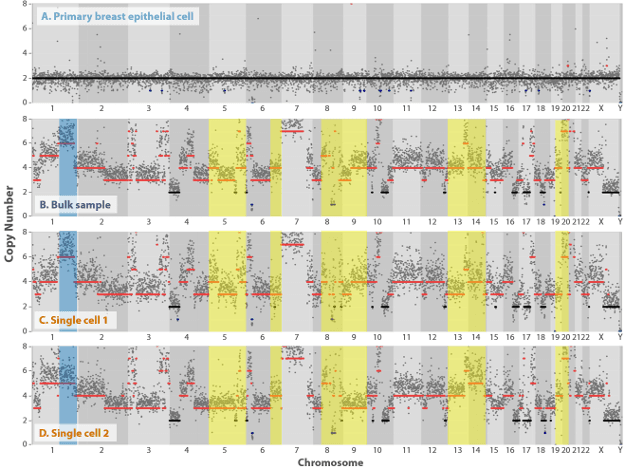
Figure 3. Bulk vs single cell CNV analysis. Compared to a normal, primary single breast epithelial cell (A), where the chromosome structure appears to be intact (diploid), the DNA from a bulk SKBR3 sample (B) displayed a high degree of copy number variation. CNV heterogeneity was clearly observed at the single cell level. In one single cell (C) the CNV pattern in chromosome 1 was similar to the bulk DNA (blue panel), whereas the degree of duplication was less in another cell (D). Differential CNV patterns were observed on the single cell level for almost every other chromosome, with the most striking examples highlighted in yellow. Given that each 500 kb window represents multiple genes, genomic differences between these individual cells are significant.
References
- Sung J, et al. CA Cancer J Clin 2021;71:209 249.
doi: 10.3322/caac.21660. - Nowell, PC. Science 1976; 194(4260):23 28.
doi: 10.1126/science.959840. - Gonzalez V, et al. bioRxiv. 2020 Nov;
doi: 10.1101/2020.11.20.391961. - West JAA, et al. Manuscript in preparation.


For more information or technical assistance: info@bioskryb.com