

Authors
ResolveDNA® Integration with Illumina DNA Prep and DNA Prep with Enrichment to Enable Single-cell Genomics.
BioSkryb Genomics has established a workflow to enable low DNA input and single-cell genomic analysis from a variety of samples. The workflow presented highlights how next generation single cell amplification, library preparation and sequencing technologies are able to obtain high quality single-cell whole genome and targeted panel data.
Introduction
Cellular heterogeneity dictates the fate of all tissues in both normal development and the pathogenesis of human disease. Defining this heterogeneity has primarily been focused on gene expression profiles in single cells1,2. While analysis is highly valuable for defining variable cell populations, the highest resolution genome data possible is required in order to provide actionable information for therapeutic selection in Oncology.
The field of DNA sequencing continues to grow at a rapid rate. However, many types of potential samples remain incompatible with the library preparation methods required for comprehensive genome analysis due to limited availability of DNA and/or cell inputs. For example, numerous studieshave demonstrated that accurate identification of genetic variation in single cells is essential for understanding the role of mutation in normal development and in disease3,4,5. This variation in individual cells can be diluted below detectable levels and missed when sampled as part of a larger bulk cell population.
Previous methodologies for whole genome amplification of single cells and picrogram (pg) quantities of DNA, such as Multiple Displacement Amplification (MDA), have not been able to provide the breadth and uniformity of genomic coverage required for robust variant detection6. With the development of Primary Template-directed Amplification (PTA)7, individual cells and low DNA inputs can now be amplified with unprecedented uniformity, providing revolutionary sequencing breadth and sensitivity.

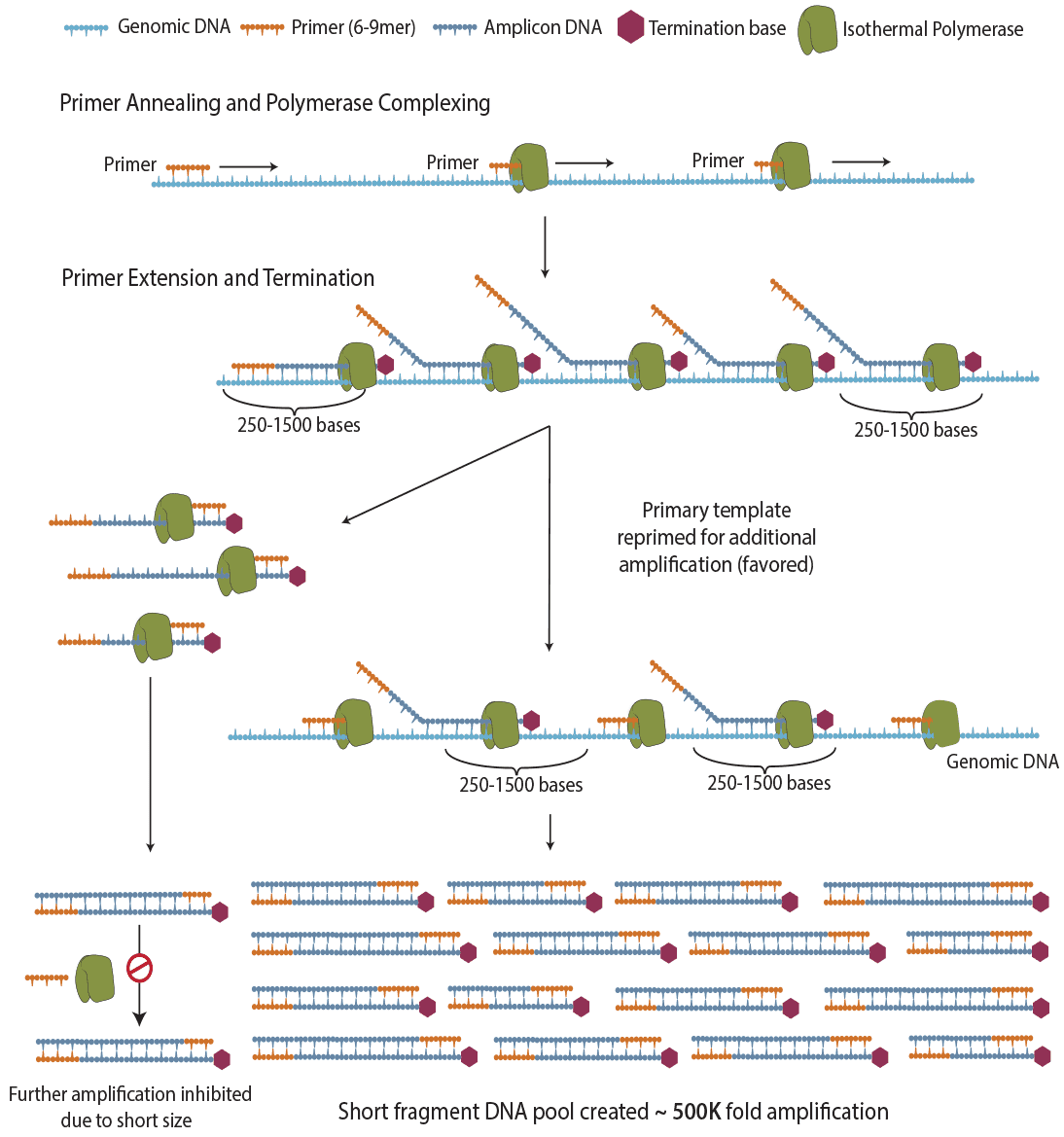
Figure 1. The principle of PTA. ResolveDNA provides unbiased amplification by utilizing random priming combined with amplicon termination to produce a true representation of original sample template. A novel approach employing proprietary nucleotides prevents the production of long amplicons, which are kinetically unfavored to be re-copied during the amplification reaction. By limiting the size of the produced amplicon, primers are re-directed to the primary template.

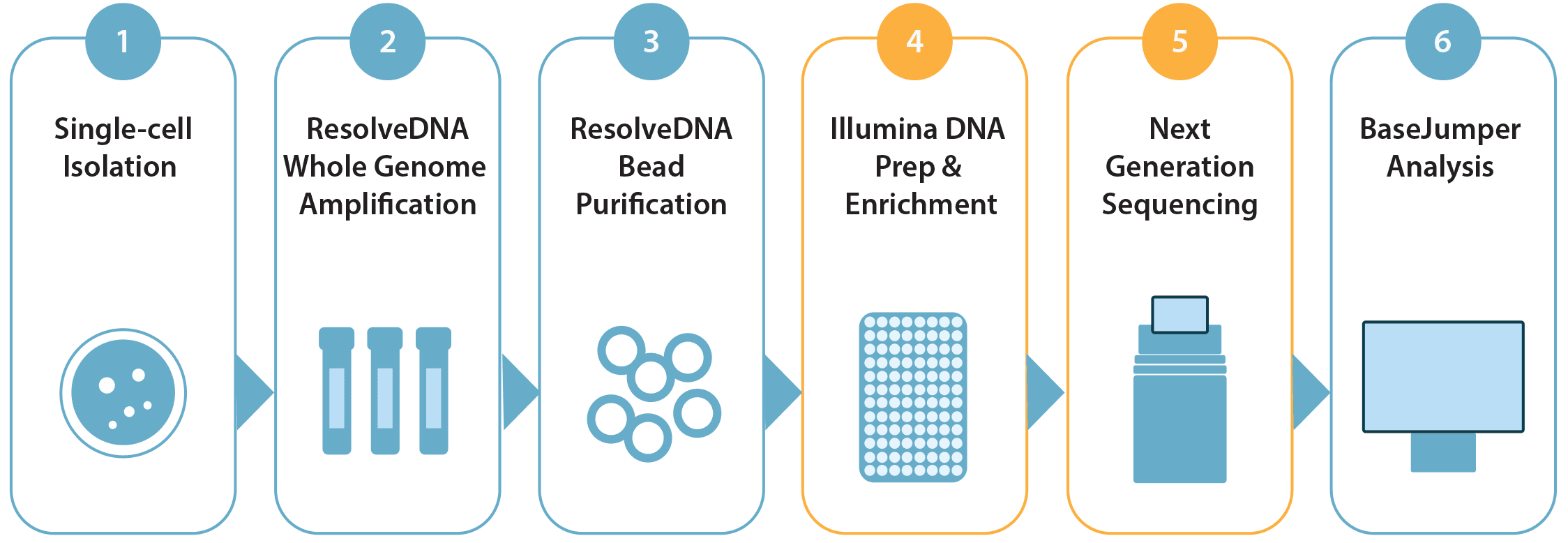
Figure 2. ResolveDNA Workflow: Isolated single cells undergo Primary Template-directed Amplification, followed by tagmentation-based library preparation, next generation sequencing, and analysis with BaseJumper™ software.
PTA utilizes isothermal amplification and proprietary termination chemistry to restrict amplicon size, preferentially redirecting random primers to the primary template (Figure 1). By limiting product amplification bias and error propagation, PTA has enabled highly accurate whole-genome and targeted analysis of a new class of samples using the Illumina DNA Prep and DNA Prep with Enrichment products. We describe here an integrated workflow for the two discrete products (Figure 2).
METHODS AND RESULTS
SAMPLE PREPARATION
Normal diploid GM12878 B-lymphoblastoid cells were subjected to PTA using ResolveDNA reagents from BioSkryb Genomics as was 50 pg or 100 pg of matched gDNA extracted from the same cells8. Individual cells were first isolated by Flourescence Activated Cell Sorting (FACS) to 96-well PCR plates at 1, 3, and 5 cells per well, with four replicates per condition. No-template control (NTC) reactions containing cell buffer only were run in parallel. Samples were incubated for 10 hours with ResolveDNA reagents. PTA products underwent 2X SPRI bead purification and total yield was determined by Qubit. Typical cell and gDNA PTA yields (Figure 3) averaged 1.7 to 4.8 ug depending on the type of input and amount used, while amplified DNA from NTC reactions were undetectable. It should also be noted that some

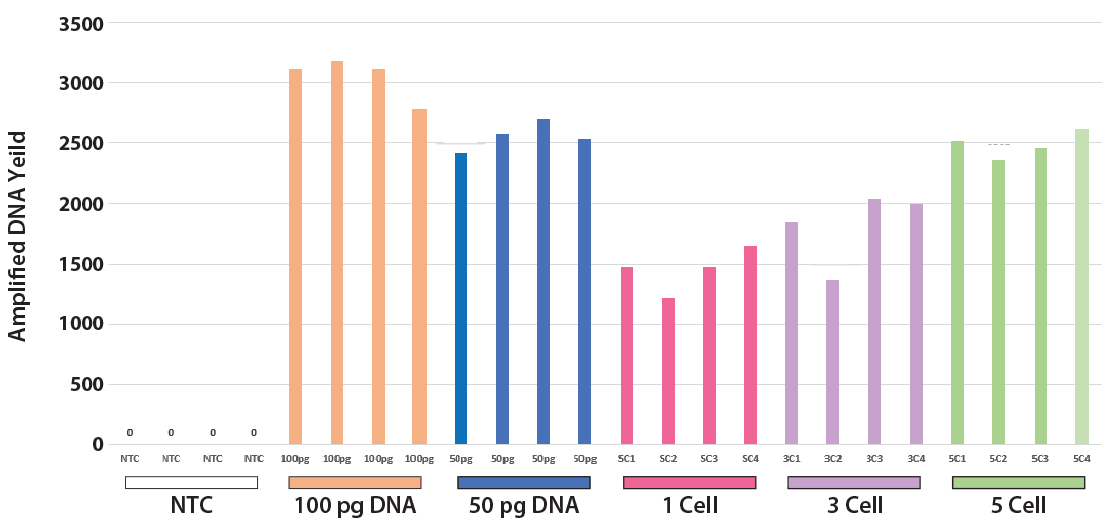
Figure 3. Single-cell and low input DNA yield. Purified DNA (100& 50 pg), along with samples containing 1, 3, and 5 cells were amplified for a period of 10 hrs, including NTC. Resulting amplified pools were purifed and quantitated by Qubit.
FACS dropout is typically observed with single cell preps and is associated with yields that are 0-25% of those from successful preps. The optimum PTA incubation time should be determined for the user’s specific cell line or primary sample. The average ploidy or total amount of genomic template can influence the PTA reaction for the input of interest. In general, diploid cells will require longer incubation times to reach yields comparable to those of polyploid cells. Higher inputs will require shorter incubation times to achieve metrics comparable to those obtained using lower inputs.
LIBRARY PREPARATION AND SEQUENCING
100 ng of purified PTA product was used as input for the Illumina DNA Prep and DNA preps with Enrichment workflows according

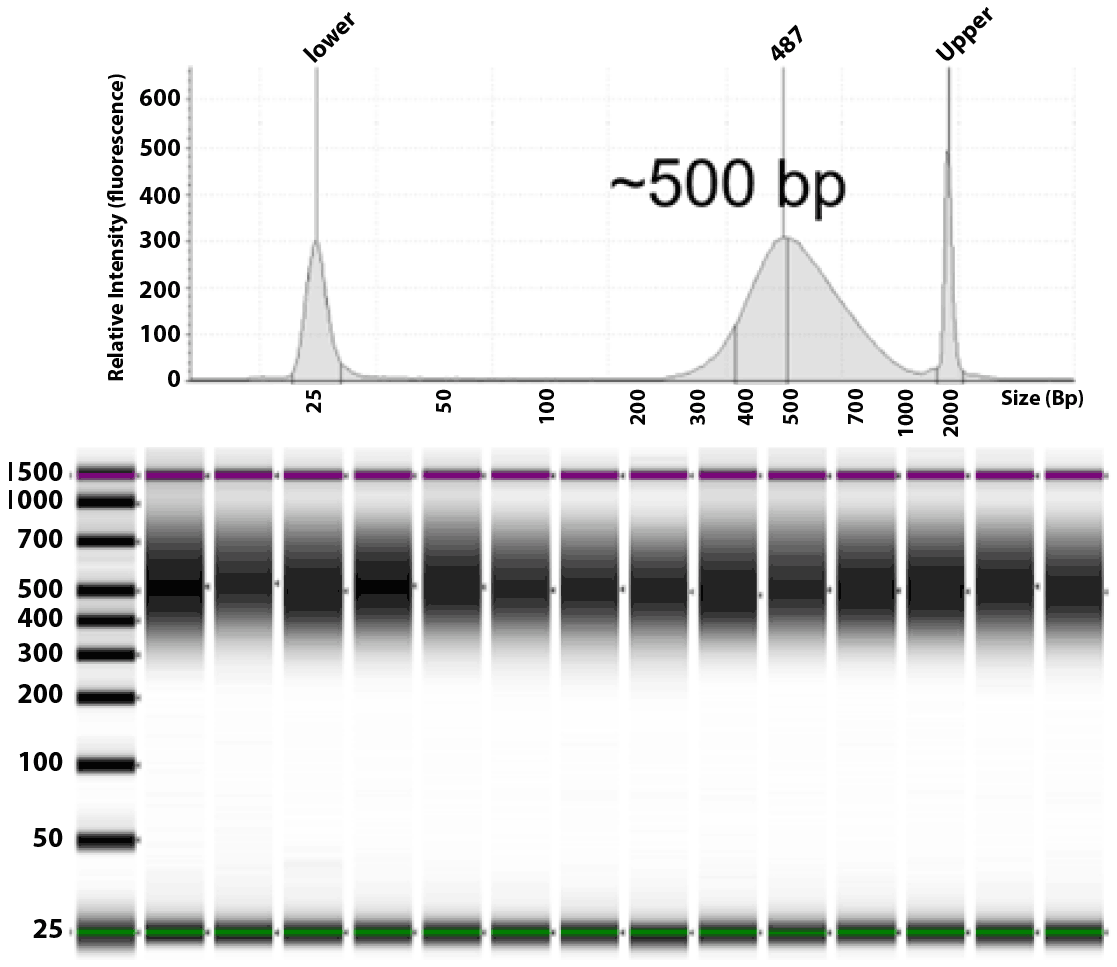
Figure 4. Library fragment size of PTA amplified products: TapeStation gel image and electropherogram sizing Illumina DNA Prep libraries derived from single GM12878 cells. Illumina DNA prep libraries prepared from ResolveDNA amplified DNA typically generate a fragment size of approximately 500 bp (~300bp insert). This pattern was reproducible across both low input DNA, single-cell and multiple (3&5) cell samples.
to the recommended conditions. The concentration of eluted whole-genome libraries averaged ~10 ng/ul for a total yield of ~300 ng, and the concentration of targeted libraries averaged ~50 ng/ul for a total yield of ~750 ng. Figure 4 highlights sizing of the DNA Prep libraries as assessed by Agilent TapeStation. This is consistent with the average expected library size of ~500bp. Sizing of DNA Prep with Enrichment libraries was confirmed to be consistent with expected sizing prior to enrichment (350bp). For enrichment, 12 libraries were pooled by mass using 250ng each and prepared with the TruSight One Expanded panel according to the recommended conditions. This panel targets ~16.5 Mb of genomic content representing ~6700 genes with broad relevance to human disease.
Inherent variability in cell state (cell cycle, chromatin-state, viability) between individual FACS-isolated cells can influence library complexity, as can subtle differences in lysis performance between individual PTA reactions. To ensure adequate genomic coverage and uniformity, each library was initially sequenced to a minimum of 2M total reads to establish its Preseq count. This is an estimation of library complexity that predicts the ability to call variants with high sensitivity and precision at the depths normally required for accurate analysis9. Only those libraries that achieved a Preseq count >3.5 E9 were used for sequencing at full depth. Both WGS and TruSight One Expanded enrichment pools were sequenced at 2X150 on NovaSeq 6000 S4 flowcells. All libraries were sequenced at recommended loading concentrations.
SEQUENCING PERFORMANCE
Mapping Metrics: For analysis of sequencing performance, WGS datasets were downsampled to 450M total reads and processed using Sentieon joint genotyping (hg38 reference) followed by variant evaluation using VQSLOD (variant quality score log-odds). TruSight One Expanded enrichments were

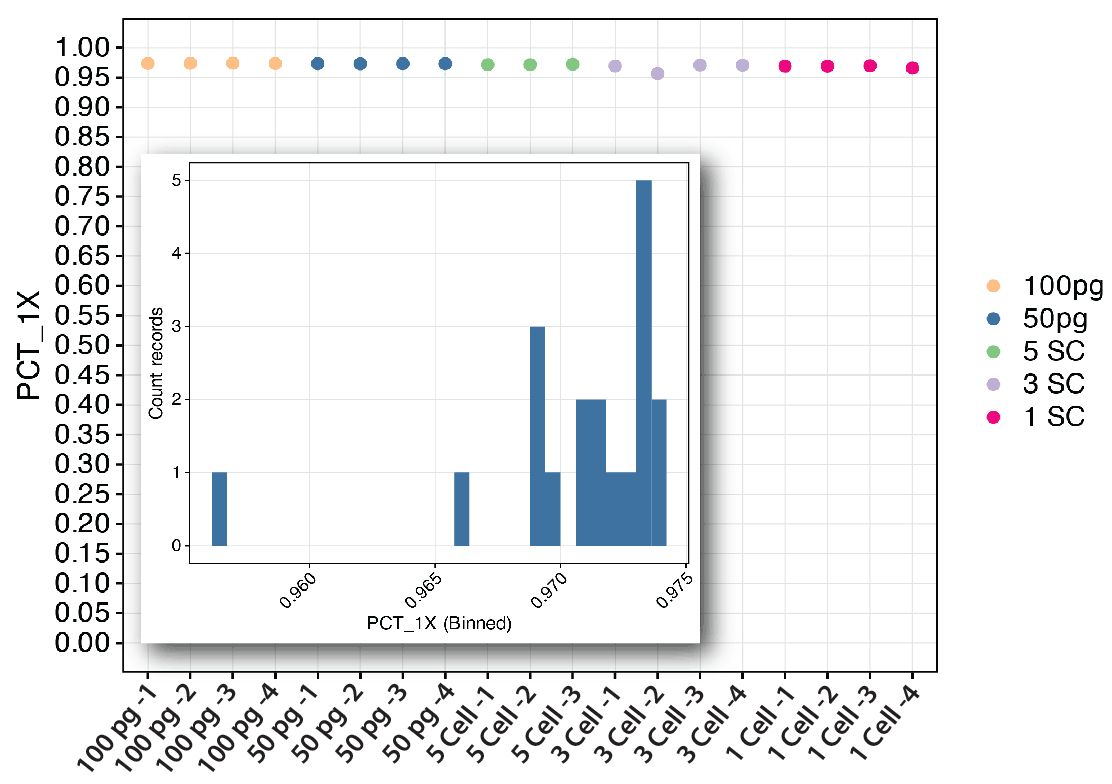
Figure 5. Whole genome coverage. Coverage was assessed over low input DNA samples (100 pg and 50 pg) as well as samples that contained 1, 3 or 5 cells/reaction. We found all samples provided greater than 95% coverage (or recovery) of the genome after ResolveDNA amplification and Illumina DNA Prep library preparation, and most were above 97% genome coverage (inset).
downsampled to 40M reads and processed using Sentieon (no joint genotyping, hg19 reference) and VQSLOD for variant evaluation.
Coverage Metrics: While mapping quality is essential to library performance, it is nonetheless of limited value if read mapping is restricted to only a portion of the genome, as has been the case with earlier whole-genome amplification technologies. To characterize the genomic and targeted coverage achieved with PTA-based libraries, high depth sequencing was employed to determine proportion of genome recovered from a range of genome material inputs. This included purified DNA samples and samples with 1, 3 and 5 cells for both WGS and targeted

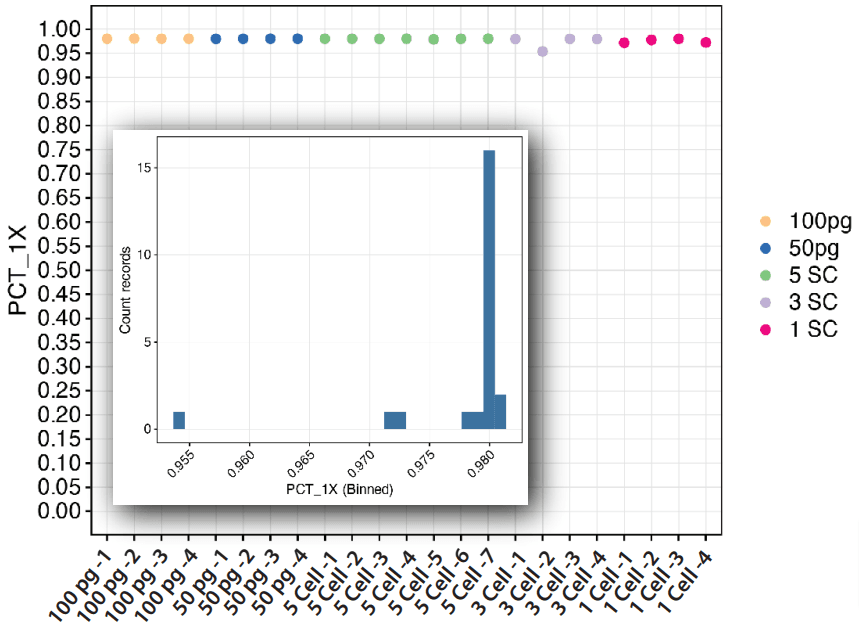
Figure 6. TruSight One Expanded genome coverage. Coverage was assessed over low input DNA samples (100 pg and 50 pg) as well as samples that contained 1, 3 or 5 cells/reaction. Similar to the WGS data, we found all samples provided greater than 95% coverage (or recovery) of the genome after ResolveDNA amplification and TruSight One Expanded enrichment, whereby most cells were above 98.0% genome coverage (inset).
panel enrichment. Overall, coverage for WGS was evenly distributed for all cell samples which contained 1, 3 or 5 cells per amplification and uniformity approached that of gDNA controls. All WGS samples (Figure 5) exceeded 95% coverage. The majority of samples had greater than 97% coverage, indicating that the overwhelming majority of DNA from the original template was recovered after ResolveDNA amplification and Illumina DNA library preparation.
Both whole-genome libraries (Figure 5) and TruSight One Expanded enrichments (Figure 6) yielded high genomic coverage. We found genomic coverage was not affected by the enrichment process, using the combined workflow of Illumina DNA Prep with enrichment library preparation and TruSight One Expanded hybrid capture. The enrichment data displayed higher genomic coverage relative to the WGS libraries with all samples having recovered greater than 95% of the genomic regions targeted by the TSO expanded panel. And the majority of samples have coverage that exceed 98%. Taken together, the data demonstrates that the performance difference between Illumina DNA Prep and Illumina DNA Prep with Enrichment
libraries is negligible. This enables unprecedented genomic coverage in both whole genome and targeted panel applications from samples which contain as few as one complete genome per reaction. The ability to capture and maintain this level of genome coverage is possible due to the combination of amplification uniformity as well as the tagmentation process which appears to minimize non-uniformity during the library preparation process. Additionally, this high coverage and uniformity translates to samples beyond GM12878, including clinical samples10.
Allelic Balance: A key factor driving the precision and sensitivity in the detection of single nucleotide variation (SNV) within single cells and low input samples is the ability to maintain strict allelic balance of heterozygous sites within the genome. Results show that allelic balance was high in amplified genomes from both low input and single cell samples (Figure 7). Analysis of allelic balance in low input, and in samples with 1, 3 or 5 cells show that the allelic balance is preserved over all samples. The data demonstrates the ResolveDNA WGA process integrated with Illumina DNA Prep library preparation system maintains a true representation of the allele frequency from both low input of bulk DNA mixtures as well as samples with a single

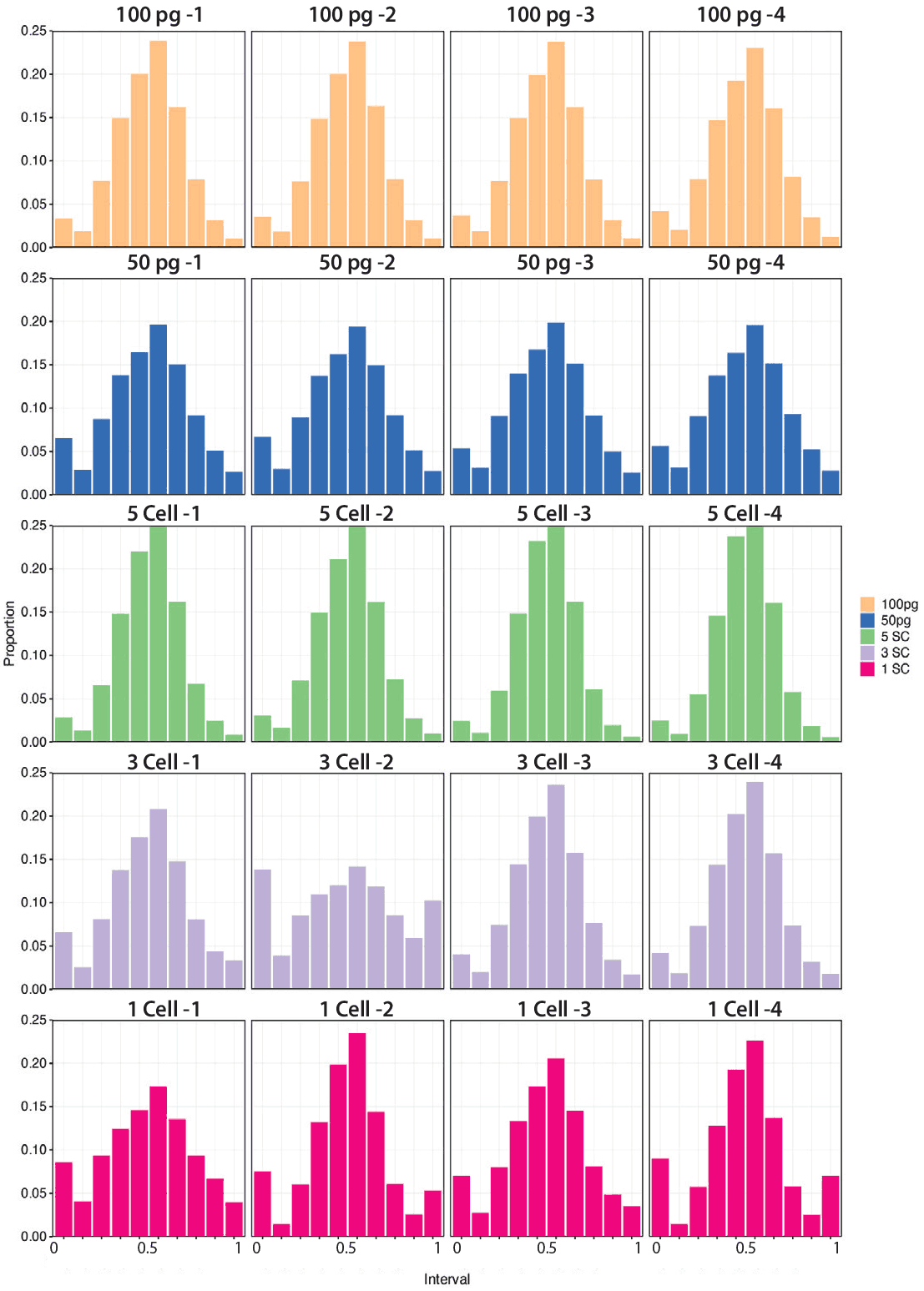
Figure 7. Allelic balance of heterozygous sites was measured in whole-genome reactions using alternate allele frequency sites with at least 2X coverage.

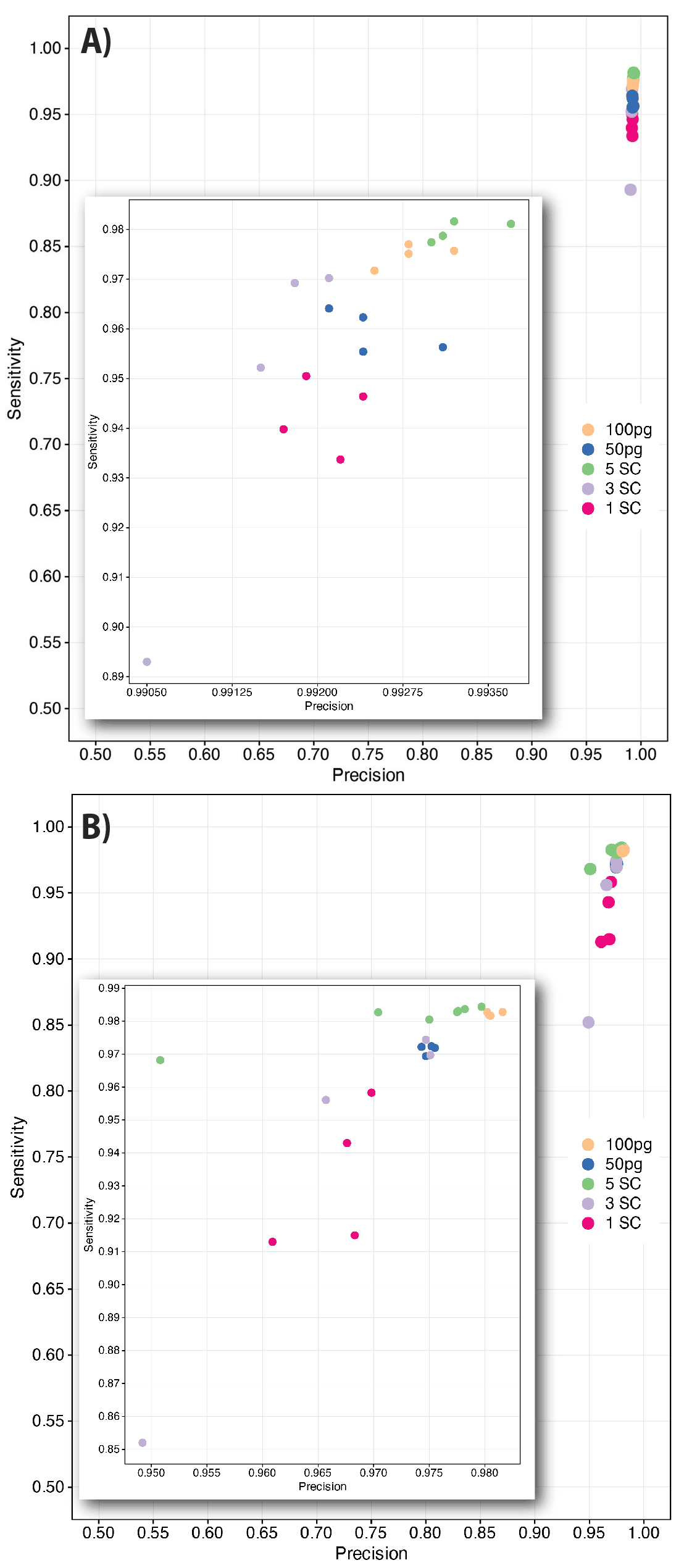
Figure 8. Precision and Sensitivity of single nucleotide variation detection was measured in both the purified DNA sample (50 & 100 pg) as well as in samples containing 1, 3 & 5 cells/ reaction. In (A) WGS data indicated precision and sensitivity exceed 99% and 95%, respectively, where in (B) precision and sensitivity of targeted enriched samples exceed 96% and 91%, respectively.
intact genome. Allelic balance was preserved in both WGS and targeted panel workflows using Illumina DNA Prep with enrichment for downstream TruSight One expanded enrichment (data not shown). This balance is a critical factor for maximizing the sensitivity and precision of allele variation across the samples analyzed in this data set.
Variant Calling Metrics: Ultimately, the performance of wholegenome amplified libraries is determined by how faithfully they represent the genome being amplified. Reference alleles for GM12878 were therefore evaluated in PTA libraries using the BaseSpace Variant Calling Assessment Tool with the Genome in a Bottle Consortium’s v.3.3.2 truth set. Figure 8 illustrates the high level of concordance observed between the PTA reactions derived from genomic DNA and the PTA reactions derived from cells.
Importantly, variant detection amongst heterozygous alleles showed limited allelic bias (Figure 7), with SNV precision and sensitivity (recall) approaching that of control libraries. Both WGS (Figure 8A) and targeted enrichment TruSight One Expanded panels (Figure 8B) display both high sensitivity and precision. WGS samples overall generated allele detection sensitivities typically greater than 90%, where the single cell had the lowest sensitivity (Figure 8A-inset) between 93-96%. Samples from pooled purified DNA and those with more than one cell ranged from 95-98% sensitivity, while precision exceeded 99% for all samples subjected to WGS analysis, downsampled to 450 million reads/sample. Similarly, samples prepared by Illumina DNA prep with Enrichment using the TSO expanded panel (Figure 8B), also demonstrated high allele variation detection sensitivity, typically greater than 90%. Again the single cells had the lowest sensitivity, but remained above 90%, and did not exceed 96%. Pooled control DNA and samples with greater than 1 cell per amplification reaction typically generated sensitivity exceeding 97% (Figure 8B-inset). All enriched samples, except one, demonstrated precision exceeding 95%. Taken together, these data demonstrate the combination of uniform amplification and transposase-based library preparation allow for exceptional genome recovery and allelic balance. This enables highly sensitive and precise single nucleotide variation detection, even from a single genome within an individual cell.
SUMMARY
Library preparation of ResolveDNA PTA products with Illumina DNA Prep and Illumina DNA Prep with Enrichment was found to be a robust solution for single cell and other low input genomic analyses requiring accurate SNV analysis. For both WGS and targeted enrichment, the unprecedented genomic recovery, coverage uniformity and allelic balance that PTA provides enables a multitude of applications. This includes cancer genomics, prenatal genetic testing, and microbiome research, with samples that would have otherwise been considered inaccessible to next-generation sequencing.
References:
- Pollen, A.A., et al., Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol, 2014. 32(10): p. 1053-8.
- Nowakowski, T.J., et al., Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science, 2017. 358(6368): p. 1318-1323.
- Navin, N.E., Delineating cancer evolution with single-cell sequencing. Sci Transl Med, 2015. 7(296): p. 296fs29.
- Xu, X., et al., Single-cell exome sequencing reveals singlenucleotide mutation characteristics of a kidney tumor. Cell, 2012. 148(5): p. 886-95.
- Alexander, J., et al., Utility of Single-Cell Genomics in Diagnostic Evaluation of Prostate Cancer. Cancer Res, 2018. 78(2): p. 348-358
- de Bourcy, C.F., et al., A quantitative comparison of single-cell whole genome amplification methods. PLoS One, 2014. 9(8): p. e105585.
- Gonzalez-Pena, V., et al., Accurate genomic variant detection in single cells with primary template-directed amplification. Proc Natl Acad Sci U S A, 2021. 118(24).
- BioSkryb Genomics, i., ResolveDNA Whole Genome Amplification Kit For high-quality single-cell and low-input DNA amplification, in www.bioskryb.com, B. Genomics, Editor. 2021, BioSkryb Genomics: Durham, NC. USA
- Daley, T. and A.D. Smith, Modeling genome coverage in single-cell sequencing. Bioinformatics, 2014. 30(22): p. 3159-65.
- Zawistowski, J., et al., Single-cell oncogenic mechanistic heterogeneity defined by PTA in primary Ductal Carcinoma In Situ, in Application note, Bioskryb Genomics, Editor. 2021, BioSkryb Genomics: www.bioskryb.com. p. 1-5.


For more information or technical assistance: info@bioskryb.com
TAS-030, 01/2022